


KAPRUVIA® is indicated for the treatment of moderate-to-severe pruritus associated with CKD in adult patients on haemodialysis. KAPRUVIA® should be restricted for in-centre haemodialysis use only.1

Mode of Action
KAPRUVIA® (difelikefalin) is the only therapy specifically licensed for treatment of moderate-to-severe CKD-aP in adult patients in-centre haemodialysis1

Key learning points
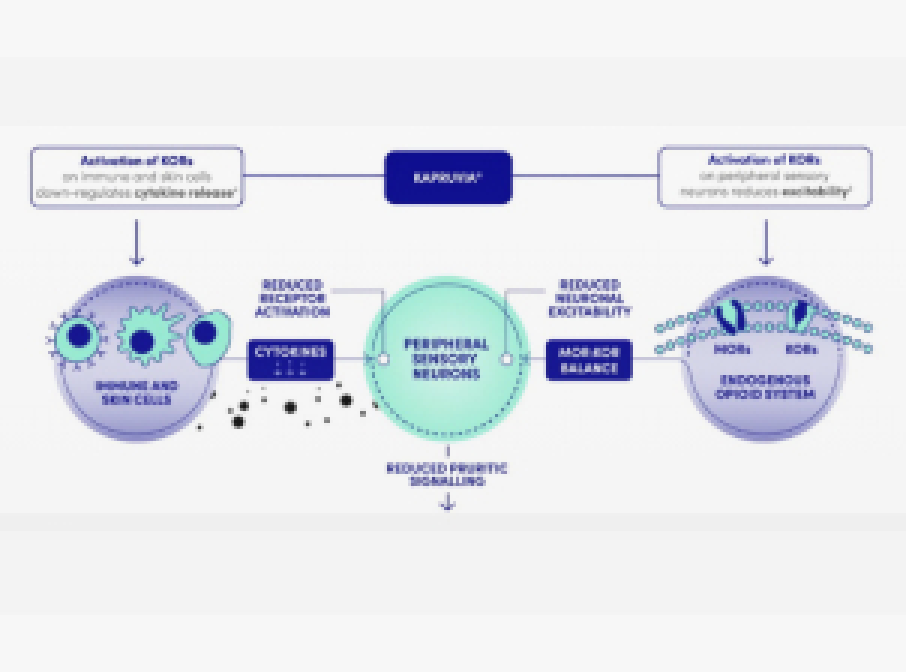
- KAPRUVIA® is a selective Kappa-opioid receptor (KOR) agonist with low central nervous system penetration1
- The anti-pruritic effects of KAPRUVIA® are considered to be due to the activation of KORs1
- KAPRUVIA® is delivered three times a week as an IV bolus at the end of in‑centre HD1
How does KAPRUVIA® work?

- KAPRUVIA® is a hydrophilic peptide with high polar surface area and charge at physiological pH1
- These physicochemical properties minimise diffusion and transport across the blood-brain barrier, limiting access to KORs in the central nervous system1,2
- The anti-pruritic and anti-inflammatory effects of KAPRUVIA® are likely due to the activation of KORs on peripheral sensory neurons and immune cells1
How should KAPRUVIA® be administered?
KAPRUVIA® is delivered as an IV bolus at the end of in-centre HD1

The recommended dose of KAPRUVIA® is 0.5 micrograms/kg dry body weight (i.e, the target post-dialysis weight)1
KAPRUVIA® is removed by the dialyser membrane and must be administered after blood is no longer circulating through the dialyser1

KAPRUVIA® is administered as an IV bolus injection into the venous line of the dialysis circuit during rinse-back or after rinse-back:1
- When given after rinse-back, at least 10 mL of sodium chloride 9 mg/mL (0.9% w/v) solution rinse-back volume should be administered after injection of KAPRUVIA®
- If the dose is given during rinse-back, no additional sodium chloride 9 mg/mL (0.9% w/v) solution is needed to flush the line

KAPRUVIA® is administered three times a week at the end of HD during rinse-back or after rinse-back1
An effect of KAPRUVIA® in reducing pruritus is expected after 2-3 weeks of treatment1
KAPRUVIA® should be continued for the duration of the dialysis, as long as there is sufficient reduction in itch in the first 12 weeks of treatment3

A fourth dose of KAPRUVIA® within 1 week is considered as an additional treatment but may be administered at the end of HD in the case of a fourth dialysis session1
No more than four doses are recommended, even if the number of dialysis treatments in a week is more than four1
Safety and efficacy of a fourth dose has not been fully established due to insufficient data1
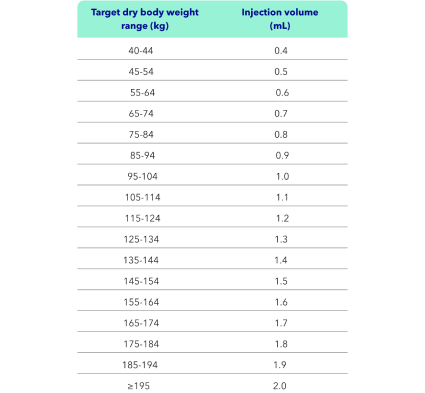
KAPRUVIA® injection volumes1
The total dose volume (mL) required from the vial should be calculated as: 0.01 × dry body weight (kg), rounded to the nearest tenth (0.1 mL). Please refer to the table.
Dosing is the same for elderly patients aged ≥65 years as for younger adult patients.
No dose adjustment is required for patients with mild or moderate hepatic impairment. KAPRUVIA® has not been studied in subjects with severe hepatic impairment and is therefore not recommended for use in this patient population.

- KAPRUVIA® should not be diluted or mixed with other medicinal products
- Vials are for single-use only; discard any unused product
- KAPRUVIA® should be a clear, colourless solution, free from particles. Inspect for any particulate matter or discolouration before administration
The safety and efficacy of KAPRUVIA® in children aged 0-17 years have not yet been established. No data are available. KAPRUVIA® has not been studied in subjects with severe hepatic impairment (National Cancer Institute Organ Dysfunction Working Group) and is therefore not recommended for use in this patient population.
KAPRUVIA® administration should be restricted for in-centre HD only and is intended for use by HCPs experienced in the diagnosis and treatment of moderate-to-severe CKD-aP. Causes of Pruritus other than chronic kidney disease should be excluded before initiating treatment with KAPRUVIA®1
Refer to the SmPC for full guidance on posology and administration
References & footnotes
Footnotes
CKD-aP, chronic kidney disease-associated Pruritus; HCP, healthcare professional; HD, haemodialysis; IV, intravenous; KORs, kappa-opioid receptors.
References
- KAPRUVIA® Summary of Product Characteristics. Available at: www.medicines.org.uk
- Lipman ZM & Yosipovitch G. Expert Opin Pharmacother 2021;22(5):549-555
- NICE (2023). Difelikefalin for treating pruritus in people having haemodialysis. Available at: https://www.nice.org.uk/guidance/TA890. Date accessed: September 2024.
▼ This medicinal product is subject to additional monitoring. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions.
UK-DFK-2300062 (v2.0) | Date of preparation: March 2025
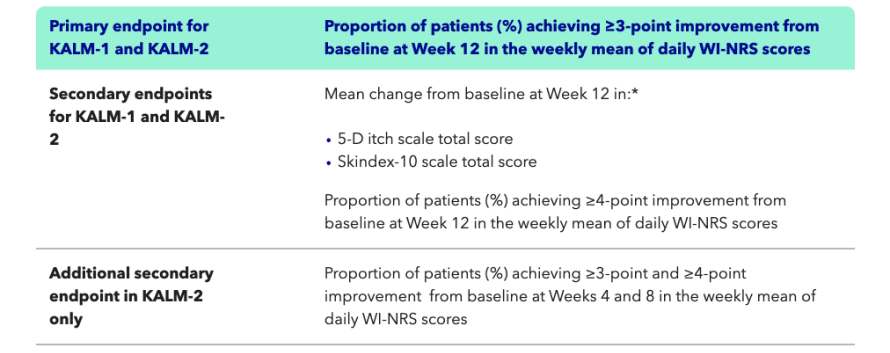
Efficacy & Safety
KALM-1: A phase III study of KAPRUVIA® (difelikefalin) vs placebo in HD patients with Pruritus1,2
Key learning points
- KALM-1 was a phase III multicentre study of KAPRUVIA® vs placebo in patients on HD with chronic kidney disease-associated Pruritus (CKD-aP)2
- More patients treated with KAPRUVIA® achieved a clinically meaningful (≥3‑point) improvement in the Worst-Itch Numerical Rating Scale (WI-NRS) vs placebo [n=189 (p<0.001)] at Week 12 (primary endpoint)1,2
- KAPRUVIA® significantly improved itch-related QoL (5-D itch and Skindex‑10) from baseline vs placebo (p<0.001) at Week 12 (secondary endpoints)1,2
The efficacy and safety of KAPRUVIA® were assessed in the KALM studies:2‑4

US multicentre study of KAPRUVIA® (n=189) vs placebo (n=189)
Completion date: April 2020

Global* multicentre study of KAPRUVIA® (n=237) vs placebo (n=236)
Completion date: March 2020
*North America (United States and Canada), Europe (Czech Republic, Germany, Great Britain, Hungary and Poland) and the Asia-Pacific region (Australia, New Zealand, South Korea and Taiwan).3
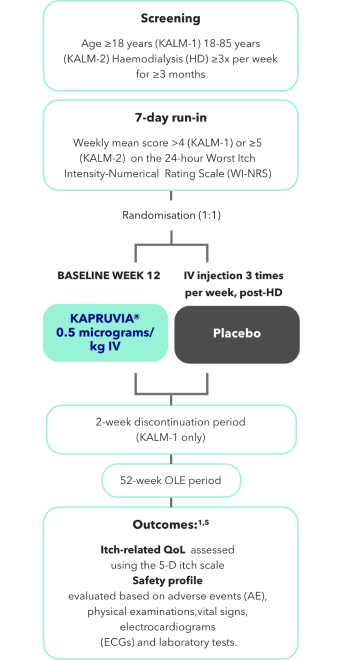
KALM-1 and KALM-2 study design2,3
KALM-1 (N=378) and KALM-2 (N=473) were two independent studies of similar design: randomised, double-blind, multicentre, placebo-controlled, phase III trials.2,3
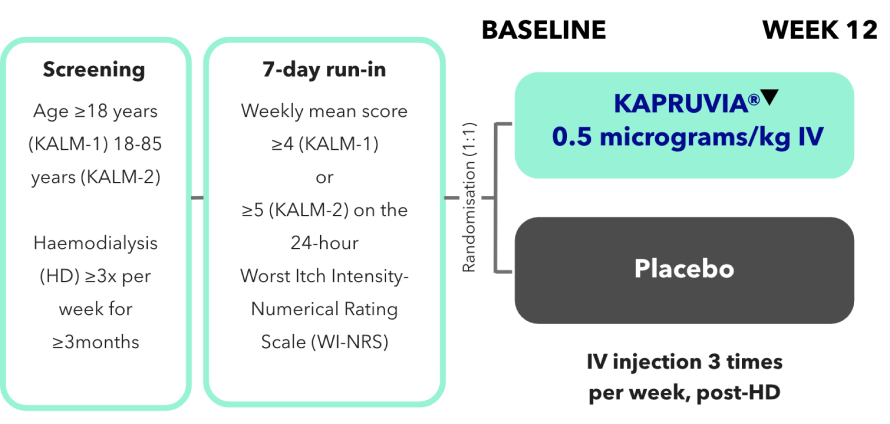
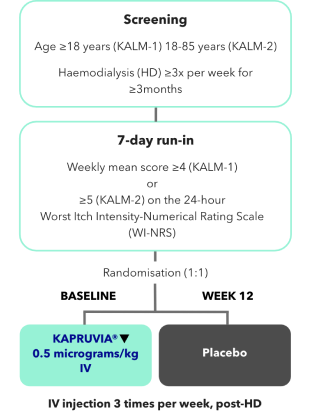
*Missing values were imputed using multiple imputation under missing-at-random assumption.2,3
In KALM-1, more patients treated with KAPRUVIA® achieved a clinically meaningful improvement in the WI-NRS vs placebo at Week 121,2
KALM-1 primary endpoint: proportion of patients with a clinically meaningful (≥3-point) improvement in WI-NRS from baseline at Week 121,2
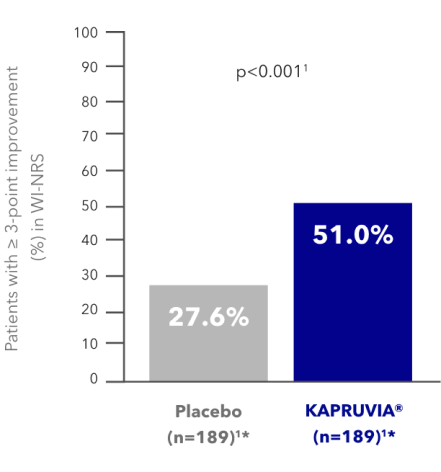
More patients treated with KAPRUVIA® achieved a clinically meaningful (≥3-point) improvement in WI-NRS vs placebo at Week 121,2

ITCH RELATED QoL 5-D ITCH
The categorical threshold of a decrease of at least 3 points was selected on the basis of a psychometric analysis of data from a previous phase 2 trial that showed that a 3-point decrease represented a clinically meaningful improvement in itch intensity in this patient population.2
Graph adapted from data in the KAPRUVIA® Summary of Product Characteristics (SmPC)1
*95% CIs are not available in the source reference for these data.1
The SmPC presents the pre-specified analysis, which included only scores during receipt of KAPRUVIA® or placebo.1
KALM-1 secondary endpoint: proportion of patients with a clinically meaningful (≥4-point) improvement in WI-NRS from baseline at Week 121,2
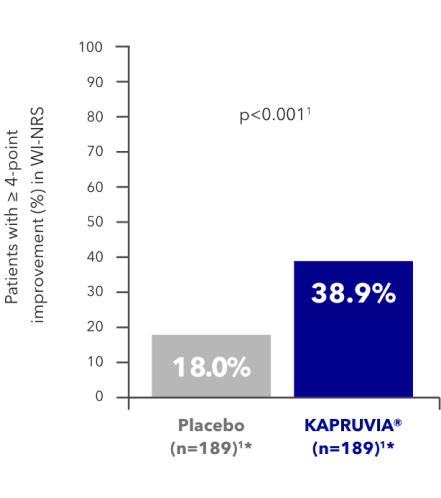
More patients treated with KAPRUVIA® achieved a clinically meaningful (≥3-point) improvement in WI-NRS vs placebo at Week 121,2
Itch intensity WI-NRS
The categorical threshold of a decrease of at least 3 points was selected on the basis of a psychometric analysis of data from a previous phase 2 trial that showed that a 3-point decrease represented a clinically meaningful improvement in itch intensity in this patient population.2
Graph adapted from data in the KAPRUVIA® SmPC1
*95% CIs are not available in the source reference for these data.1
The SmPC presents the pre-specified analysis, which included only scores during receipt of KAPRUVIA® or placebo.1
Further data for the KALM-1 study has been published in the New England Journal of Medicine, Fishbane S, et al. 2020;382:222-232.2
In KALM-1, KAPRUVIA® significantly improved itch-related QoL vs placebo at Week 121,2
KALM-1 secondary endpoint: impact of KAPRUVIA® on 5-D itch QoL scores in HD patients with CKD-aP at Week 121,2
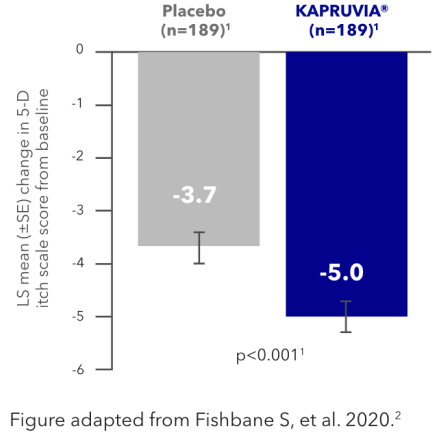
KAPRUVIA® significantly improved itch-related QoL (5-D itch) from baseline vs placebo (p<0.001) at Week 121,2

Itch intensity WI-NRS
LS mean change from baseline in 5-D itch (95% CI) KAPRUVIA® -5.0 (-5.7, -4.4), Placebo -3.7 (-4.4, -3.1)4
KALM-1 secondary endpoint: impact of KAPRUVIA® on Skindex-10 QoL scores in patients with CKD-aP at Week 121,2
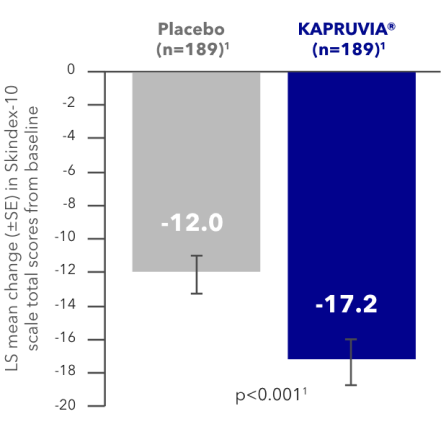
Patients treated with KAPRUVIA® achieved significantly greater mean changes from baseline to Week 12 in itch-related QoL (Skindex-10 total score) vs placebo1,2

ITCH-RELATED QoL SKINDEX‑10
LS mean change from baseline in Skindex-10 total score (95% CI) KAPRUVIA® -17.2 (-19.6, -14.7), Placebo -12.0 (-14.5, -9.6)4
Figure adapted from Fishbane S, et al. 2020.2
KAPRUVIA® safety profile
In the KALM-1 study, 68.8% (n=130/189) of patients in the KAPRUVIA® group and 62.2% (n=117/188) in the placebo group experienced adverse events (AEs). 15 patients (7.9%) in the KAPRUVIA® group discontinued treatment due to adverse events vs nine patients (4.8%) in the placebo group.2
The most common adverse reactions of KAPRUVIA® treatment are somnolence (1.1%), paraesthesia (including hypoesthesia, paraesthesia oral and hypoesthesia oral) (1.1%), dizziness (0.9%), headache (0.6%), nausea (0.7%), vomiting (0.7%), diarrhoea (0.2%) and mental status changes (including confusional states) (0.3%). Most of these events are mild or moderate in severity. Refer to the KAPRUVIA® SmPC for more information.1
References & footnotes
Footnotes
AE, adverse event; CI, confidence interval; CKD-aP, chronic kidney disease-associated Pruritus; HD, haemodialysis; KORs, kappa-opioid receptors; LS, least squares; QoL, quality of life; SE, standard error; SmPC, Summary of Product Characteristics; WI-NRS, Worst-Itch Numerical Rating Scale.
References
- KAPRUVIA® Summary of Product Characteristics. Available at: www.medicines.org.uk.
- Fishbane S, et al. N Engl J Med 2020;382(3):222-232 (+ suppl).
- Wooldridge T, et al. Efficacy and safety of difelikefalin for moderate-to-severe chronic kidney disease-associated pruritus: a global phase 3 study in hemodialysis patients (KALM-2) (FR-OR24). Presented at 2020 Kidney Week, virtual, 19-25 October 2020. Available at: caratherapeutics.com. Accessed September 2023.
- Topf J, et al. Kidney Med 2022;4(8):100512 (+ suppl).
- Fishbane S, et al. Kidney Med 2022;4(8):100513.
- Weiner D, et al. Kidney Med 2022;4(10):100542.
▼ This medicinal product is subject to additional monitoring. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions.
UK-DFK-2300066 (v2.0) | Date of preparation: March 2025
Efficacy & Safety
KALM-1: A phase III study of KAPRUVIA® (difelikefalin) vs placebo in HD patients with Pruritus1,2
Key learning points
- KALM-1 was a phase III multicentre study of KAPRUVIA® vs placebo in patients on HD with chronic kidney disease-associated Pruritus (CKD-aP)2
- More patients treated with KAPRUVIA® achieved a clinically meaningful (≥3‑point) improvement in the Worst-Itch Numerical Rating Scale (WI-NRS) vs placebo [n=189 (p<0.001)] at Week 12 (primary endpoint)1,2
- KAPRUVIA® significantly improved itch-related QoL (5-D itch and Skindex‑10) from baseline vs placebo (p<0.001) at Week 12 (secondary endpoints)1,2
The efficacy and safety of KAPRUVIA® were assessed in the KALM studies:2‑4
US multicentre study of KAPRUVIA® (n=189) vs placebo (n=189)
Completion date: April 2020
Global* multicentre study of KAPRUVIA® (n=237) vs placebo (n=236)
Completion date: March 2020
*North America (United States and Canada), Europe (Czech Republic, Germany, Great Britain, Hungary and Poland) and the Asia-Pacific region (Australia, New Zealand, South Korea and Taiwan).3
KALM-1 and KALM-2 study design2,3
KALM-1 (N=378) and KALM-2 (N=473) were two independent studies of similar design: randomised, double-blind, multicentre, placebo-controlled, phase III trials.2,3
*Missing values were imputed using multiple imputation under missing-at-random assumption.2,3
In KALM-1, more patients treated with KAPRUVIA® achieved a clinically meaningful improvement in the WI-NRS vs placebo at Week 121,2
KALM-1 primary endpoint: proportion of patients with a clinically meaningful (≥3-point) improvement in WI-NRS from baseline at Week 121,2
More patients treated with KAPRUVIA® achieved a clinically meaningful (≥3-point) improvement in WI-NRS vs placebo at Week 121,2
ITCH RELATED QoL 5-D ITCH
The categorical threshold of a decrease of at least 3 points was selected on the basis of a psychometric analysis of data from a previous phase 2 trial that showed that a 3-point decrease represented a clinically meaningful improvement in itch intensity in this patient population.2
Graph adapted from data in the KAPRUVIA® Summary of Product Characteristics (SmPC)1
*95% CIs are not available in the source reference for these data.1
The SmPC presents the pre-specified analysis, which included only scores during receipt of KAPRUVIA® or placebo.1
KALM-1 secondary endpoint: proportion of patients with a clinically meaningful (≥4-point) improvement in WI-NRS from baseline at Week 121,2
More patients treated with KAPRUVIA® achieved a clinically meaningful (≥3-point) improvement in WI-NRS vs placebo at Week 121,2
Itch intensity WI-NRS
The categorical threshold of a decrease of at least 3 points was selected on the basis of a psychometric analysis of data from a previous phase 2 trial that showed that a 3-point decrease represented a clinically meaningful improvement in itch intensity in this patient population.2
Graph adapted from data in the KAPRUVIA® SmPC1
*95% CIs are not available in the source reference for these data.1
The SmPC presents the pre-specified analysis, which included only scores during receipt of KAPRUVIA® or placebo.1
Further data for the KALM-1 study has been published in the New England Journal of Medicine, Fishbane S, et al. 2020;382:222-232.2
In KALM-1, KAPRUVIA® significantly improved itch-related QoL vs placebo at Week 121,2
KALM-1 secondary endpoint: impact of KAPRUVIA® on 5-D itch QoL scores in HD patients with CKD-aP at Week 121,2
KAPRUVIA® significantly improved itch-related QoL (5-D itch) from baseline vs placebo (p<0.001) at Week 121,2
Itch intensity WI-NRS
LS mean change from baseline in 5-D itch (95% CI) KAPRUVIA® -5.0 (-5.7, -4.4), Placebo -3.7 (-4.4, -3.1)4
KALM-1 secondary endpoint: impact of KAPRUVIA® on Skindex-10 QoL scores in patients with CKD-aP at Week 121,2
Patients treated with KAPRUVIA® achieved significantly greater mean changes from baseline to Week 12 in itch-related QoL (Skindex-10 total score) vs placebo1,2
ITCH-RELATED QoL SKINDEX‑10
LS mean change from baseline in Skindex-10 total score (95% CI) KAPRUVIA® -17.2 (-19.6, -14.7), Placebo -12.0 (-14.5, -9.6)4
Figure adapted from Fishbane S, et al. 2020.2
KAPRUVIA® safety profile
In the KALM-1 study, 68.8% (n=130/189) of patients in the KAPRUVIA® group and 62.2% (n=117/188) in the placebo group experienced adverse events (AEs). 15 patients (7.9%) in the KAPRUVIA® group discontinued treatment due to adverse events vs nine patients (4.8%) in the placebo group.2
The most common adverse reactions of KAPRUVIA® treatment are somnolence (1.1%), paraesthesia (including hypoesthesia, paraesthesia oral and hypoesthesia oral) (1.1%), dizziness (0.9%), headache (0.6%), nausea (0.7%), vomiting (0.7%), diarrhoea (0.2%) and mental status changes (including confusional states) (0.3%). Most of these events are mild or moderate in severity. Refer to the KAPRUVIA® SmPC for more information.1
References & footnotes
Footnotes
AE, adverse event; CI, confidence interval; CKD-aP, chronic kidney disease-associated Pruritus; HD, haemodialysis; KORs, kappa-opioid receptors; LS, least squares; QoL, quality of life; SE, standard error; SmPC, Summary of Product Characteristics; WI-NRS, Worst-Itch Numerical Rating Scale.
References
- KAPRUVIA® Summary of Product Characteristics. Available at: www.medicines.org.uk.
- Fishbane S, et al. N Engl J Med 2020;382(3):222-232 (+ suppl).
- Wooldridge T, et al. Efficacy and safety of difelikefalin for moderate-to-severe chronic kidney disease-associated pruritus: a global phase 3 study in hemodialysis patients (KALM-2) (FR-OR24). Presented at 2020 Kidney Week, virtual, 19-25 October 2020. Available at: caratherapeutics.com. Accessed September 2023.
- Topf J, et al. Kidney Med 2022;4(8):100512 (+ suppl).
- Fishbane S, et al. Kidney Med 2022;4(8):100513.
- Weiner D, et al. Kidney Med 2022;4(10):100542.
▼ This medicinal product is subject to additional monitoring. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions.
UK-DFK-2300066 (v2.0) | Date of preparation: March 2025
Efficacy & Safety
KALM-2: A phase III study of KAPRUVIA® (difelikefalin) vs placebo in HD patients with Pruritus1,2
Key learning points
- KALM-2 was a global multicentre study on the effect of KAPRUVIA® vs placebo for patients on HD with chronic kidney disease‑associated Pruritus (CKD-aP)2
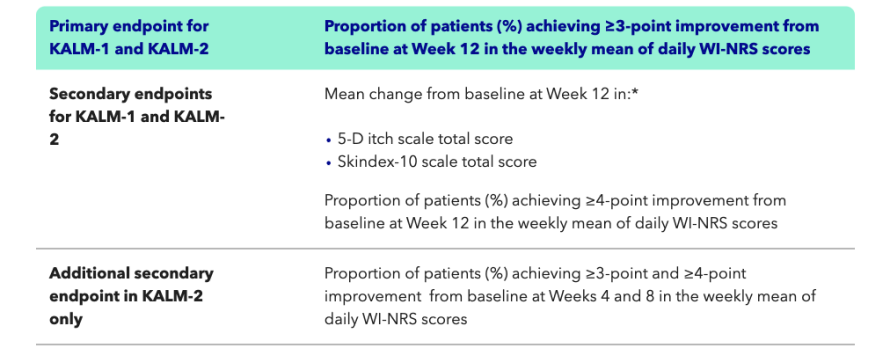
- In KALM-2, KAPRUVIA® helped more patients to achieve a clinically meaningful (≥3-point) improvement in the Worst-Itch Numerical Rating Scale (WI-NRS) vs placebo [n=237 (p=0.02)] at Week 12 (primary endpoint)1,2
The efficacy and safety of KAPRUVIA® were assessed in the KALM studies:2‑4
US multicentre study of KAPRUVIA® (n=189) vs placebo (n=189)
Completion date: April 2020
Global* multicentre study of KAPRUVIA® (n=237) vs placebo (n=236)
Completion date: March 2020
*North America (United States and Canada), Europe (Czech Republic, Germany, Great Britain, Hungary and Poland) and the Asia-Pacific region (Australia, New Zealand, South Korea and Taiwan).2
KALM-1 and KALM-2 study design2,3
KALM-1 (N=378) and KALM-2 (N=473) were two independent studies of similar design: randomised, double-blind, multicentre, placebo-controlled, phase III trials.2,3
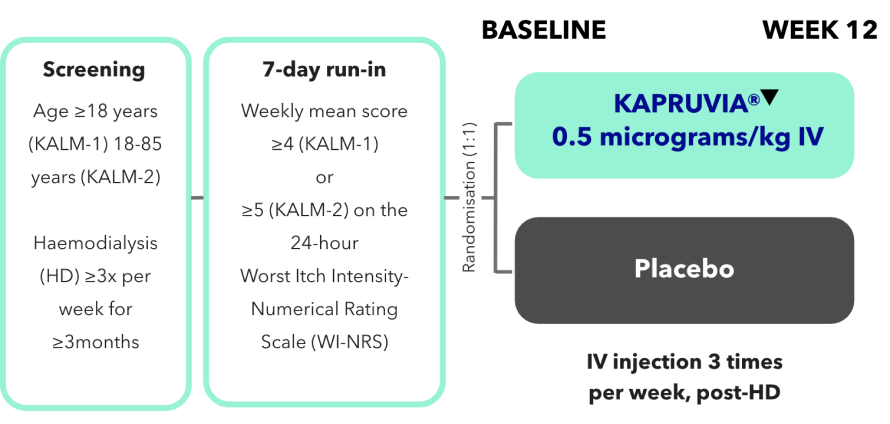
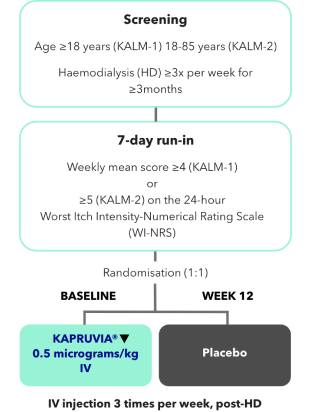

*Missing values were imputed using multiple imputation under missing-at-random assumption.2,3
In KALM-2, more patients treated with KAPRUVIA® achieved a clinically meaningful improvement in the WI-NRS vs placebo at Week 121,2,4
KALM-2 primary endpoint: proportion of patients with a clinically meaningful (≥3-point) improvement in WI-NRS from baseline at Week 121,2,4
KAPRUVIA® helped more patients to achieve a clinically meaningful (≥3-point) improvement in WI-NRS vs placebo at Week 121,2,4
Itch intensity WI-NRS
The categorical threshold of a decrease of at least 3 points was selected on the basis of a psychometric analysis of data from a previous phase 2 trial that showed that a 3-point decrease represented a clinically meaningful improvement in itch intensity in this patient population.2
Graph adapted from data in the KAPRUVIA® Summary of Product Characteristics (SmPC)1
*95% CIs are not available in the source reference for these data.1
The SmPC presents the pre-specified analysis, which included only scores during receipt of KAPRUVIA® or placebo.1
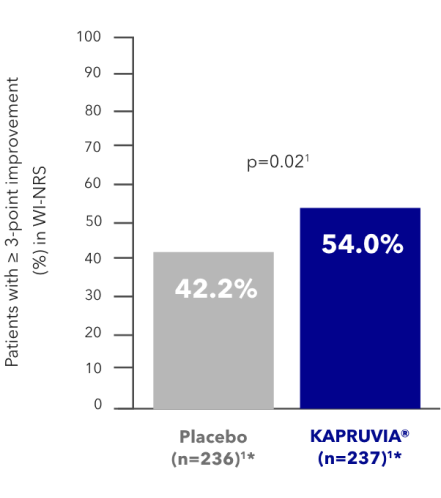
KALM-2 secondary endpoint: proportion of patients with a clinically meaningful (≥4-point) improvement in WI-NRS from baseline at Week 121,2,4
KAPRUVIA® helped more patients to achieve a clinically meaningful (≥4-point) improvement in WI-NRS vs placebo at Week 121,2,4
Itch intensity WI-NRS
The categorical threshold of a decrease of at least 3 points was selected on the basis of a psychometric analysis of data from a previous phase 2 trial that showed that a 3-point decrease represented a clinically meaningful improvement in itch intensity in this patient population.2
Graph adapted from data in the KAPRUVIA® SmPC1
*95% CIs are not available in the source reference for these data.1
The SmPC presents the pre-specified analysis, which included only scores during receipt of KAPRUVIA® or placebo.1
The effect of KAPRUVIA® on itch‑related QoL vs placebo at Week 121,2,4
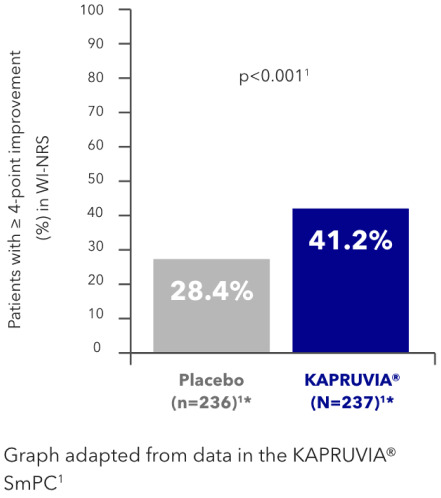
KALM-2 secondary endpoint: impact of KAPRUVIA® on 5-D itch QoL scores in HD patients with CKD-aP at Week 121,2,4
Patients treated with KAPRUVIA® showed a reduction in total 5-D itch scale score from baseline of -4.9 at Week 12. The improvement was not significantly different from placebo (-3.8; p=not applicable)1,2,4*
Itch-related QoL 5-D itch
Figure developed from data in Topf, et al. 2022. Supplementary appendix.4
*Nominal p-value not considered influential based on sequential statistical analysis.4 P-value was not tested based on the hierarchical testing order.1
KALM-2 secondary endpoint: impact of KAPRUVIA® on Skindex-10 QoL scores in patients with CKD-aP at Week 121,2,4
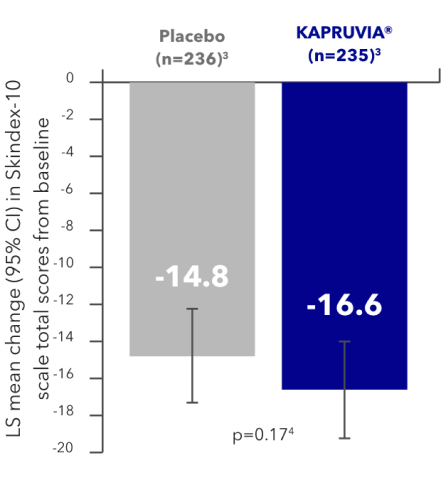
Patients treated with KAPRUVIA® showed improved itch-related QoL (total Skindex-10 score) from baseline at Week 12. The improvement failed to reach significance vs placebo (p=0.17)1,2,4
ITCH-RELATED QoL SKINDEX‑10
Figure developed from data in Topf, et al. 2022. Supplementary appendix.4
Study limitations: missing data were imputed using multiple imputation under missing-at-random assumption.2,4
KAPRUVIA® safety profile
In the KALM-2 study, 68.1% (n=160/235) of patients in the KAPRUVIA® group and 61.4% (n=145/236) in the placebo group experienced adverse events (AEs). 13 patients (5.5%) in the KAPRUVIA® group discontinued treatment due to AEs vs 8 patients (3.4%) in the placebo group.2
The most common adverse reactions of KAPRUVIA® treatment are somnolence (1.1%), paraesthesia (including hypoesthesia, paraesthesia oral and hypoesthesia oral) (1.1%), dizziness (0.9%), headache (0.6%), nausea (0.7%), vomiting (0.7%), diarrhoea (0.2%) and mental status changes (including confusional states) (0.3%). Most of these events are mild or moderate in severity. Refer to the KAPRUVIA® SmPC for more information.1
References & footnotes
Footnotes
AE, adverse event; CI, confidence interval; CKD-aP, chronic kidney disease-associated Pruritus; HD, haemodialysis; LS, least squares; QoL, quality of life; SmPC, Summary of Product Characteristics; WI-NRS, Worst-Itch Numerical Rating Scale.
References
- KAPRUVIA® Summary of Product Characteristics. Available at: www.medicines.org.uk.
- Wooldridge T, et al. Efficacy and safety of difelikefalin for moderate-to-severe chronic kidney disease-associated Pruritus: a global phase 3 study in hemodialysis patients (KALM-2) (FR-OR24). Presented at 2020 Kidney Week, virtual, 19-25 October 2020. Available at: caratherapeutics.com. Accessed September 2023.
- Fishbane S, et al. N Engl J Med 2020;382(3):222-232 (+ suppl).
- Topf J, et al. Kidney Med 2022;4(8):100512 (+ suppl).
▼ This medicinal product is subject to additional monitoring. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions.
UK-DFK-2300067 (v2.0) | Date of preparation: March 2025
Safety & Efficacy
KALM-1 and KALM-2 post-hoc pooled analysis2
Key learning points
- The KALM-1 and KALM-2 pooled analysis showed that more patients achieved a ≥3-point improvement in Worst-Itch Numerical Rating Scale (WI-NRS) score with KAPRUVIA® vs placebo (p<0.001) as early as Week 2 and this continued up to Week 122
- In patients switching from placebo to KAPRUVIA® at the end of the double-blind phase, an improvement in the 5-D itch score was observed after 4 weeks of treatment and was maintained up to 52 weeks.2
Efficacy and safety of KAPRUVIA® were demonstrated in the KALM studies1,3,4
KALM-1 (N=378) and KALM-2 (N=473) were two independent studies of similar design: randomised, double-blind, multicentre, placebo-controlled, phase III trials.1,3,4
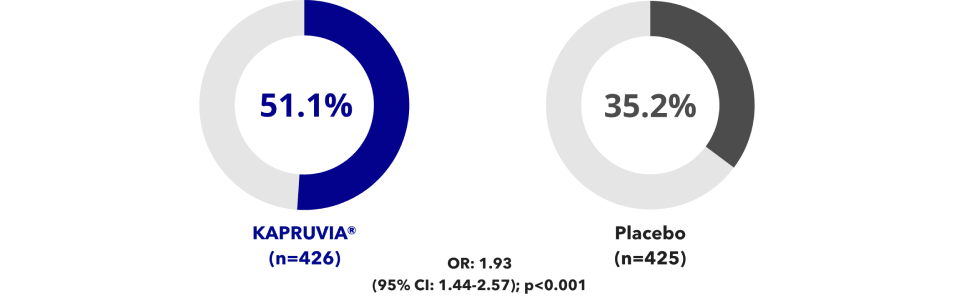
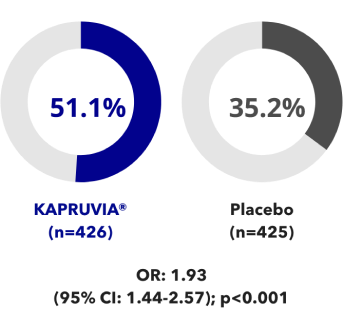
Data from KALM-1 and KALM-2 were pooled in a post-hoc analysis2
Data from a total of 851 patients from KALM-1 and KALM-2 were combined in the pooled analysis from baseline to Week 12 (KAPRUVIA®: n=426; placebo: n=425).
340 patients from KALM-1 and KALM-2 who received KAPRUVIA® in the double-blind, randomised, placebo-controlled phase proceeded to the OLE, alongside 372 patients who switched from placebo to KAPRUVIA®.2

Effects of KAPRUVIA® on itch intensity from baseline up to Week 12 vs placebo2
More patients achieved a ≥3‑point improvement in WI‑NRS score with KAPRUVIA® at Week 122
From Week 1 through to Week 12 more patients treated with KAPRUVIA® achieved a ≥3-point improvement in WI-NRS score2
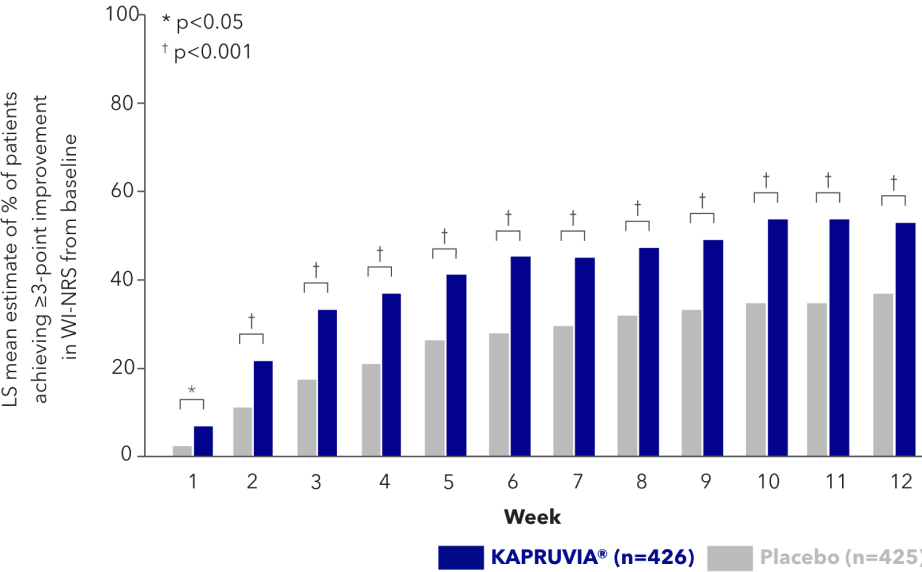
Study limitations: this is a post-hoc analysis of pooled data from two studies, KALM-1 and KALM-2. The WI‑NRS is not used in routine clinical care of dialysis patients; therefore, findings may not reflect the real-world effectiveness of KAPRUVIA®. Patient-reported outcomes such as the WI‑NRS are susceptible to placebo effects, which likely impacted the observed treatment effects.2
Effects of KAPRUVIA® on 5-D itch score up to Week 522

Figure adapted from Topf J, et al. 20222
*p-values for the significance of this analysis were not available in the supporting publication.
†Graph does not show the 2-week discontinuation process for those patients who did not enter the OLE.2
Study limitations: the 5-D itch score is not used in routine clinical care of dialysis patients; therefore, findings may not reflect the real-world effectiveness of KAPRUVIA®.2 Analyses of significance of any findings in the KALM-2 study were nominal and not considered influential based on sequential statistical analysis.1,4
KAPRUVIA® safety profile
The most common adverse reactions of KAPRUVIA® treatment are somnolence (1.1%), paraesthesia (including hypoesthesia, paraesthesia oral and hypoesthesia oral) (1.1%), dizziness (0.9%), headache (0.6%), nausea (0.7%), vomiting (0.7%), diarrhoea (0.2%) and mental status changes (including confusional states) (0.3%). Most of these events are mild or moderate in severity.
Refer to the KAPRUVIA® Summary of Product Characteristics for more information.1
References & footnotes
Footnotes
CI, confidence interval; CKD-aP, chronic kidney disease-associated Pruritus; ECG, electrocardiogram; IV, intravenous; LS, least squares; OLE, open-label extension; OR, odds ratio; QoL, quality of life; WI-NRS, Worst-Itch Intensity Numerical Rating Scale.
References
- KAPRUVIA® Summary of Product Characteristics. Available at: www.medicines.org.uk.
- Topf J, et al. Kidney Med 2022;4(8):100512 (+ suppl).
- Fishbane S, et al. N Engl J Med 2020;382(3):222-232 (+ suppl).
- Wooldridge T, et al. Efficacy and safety of difelikefalin for moderate-to-severe chronic kidney disease-associated pruritus: a global phase 3 study in hemodialysis patients (KALM-2) (FR-OR24). Presented at 2020 Kidney Week, virtual, 19-25 October 2020. Available at: caratherapeutics.com. Accessed December 2023.
- Fishbane S, et al. Kidney Med 2022;4(8):100513.
▼ This medicinal product is subject to additional monitoring. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions.
UK-DFK-2300070 (v2.0) | Date of preparation: March 2025
Safety & Efficacy
KAPRUVIA® (difelikefalin) is generally well tolerated1,2
Key learning points
- KAPRUVIA® has been shown in clinical trials to be generally well tolerated1
- The most common (≥1/100 to <1/10) adverse events (AEs) attributed to KAPRUVIA® in haemodialysis (HD) patients are somnolence and paraesthesia1
Summary of the safety profile1

Somnolence1
Somnolence was reported as a treatment-emergent adverse event (TEAE) in 2.2% of subjects randomised to KAPRUVIA®. The vast majority of these events were mild or moderate in severity. In 0.3% of patients, somnolence led to discontinuation of treatment with KAPRUVIA®. Somnolence was reported as a serious adverse event (SAE) in <0.1% of patients treated with KAPRUVIA®

Dizziness1
Dizziness was reported as a TEAE in 7.9% of subjects randomised to KAPRUVIA®. The vast majority of these events were mild or moderate in severity. In 0.5% of patients, dizziness led to discontinuation of treatment with KAPRUVIA®. Dizziness was reported as an SAE in 0.5% of patients treated with KAPRUVIA®

Mental status change1*
Mental status change (including confusional state) was reported as a TEAE in 4.4% of patients randomised to KAPRUVIA®. The majority of these events were mild or moderate in severity. Mental status changes were reported as an SAE in 2.2% of patients treated with KAPRUVIA®
Concurrent administrations of medicinal products such as sedating antihistamines, opioid analgesics or other central nervous system (CNS) depressants may increase the likelihood of dizziness and somnolence.1
*Mental status changes included MedDRA preferred terms of confusional state and mental status changes.
Adverse reactions attributed to treatment with KAPRUVIA® in patients on HD:1
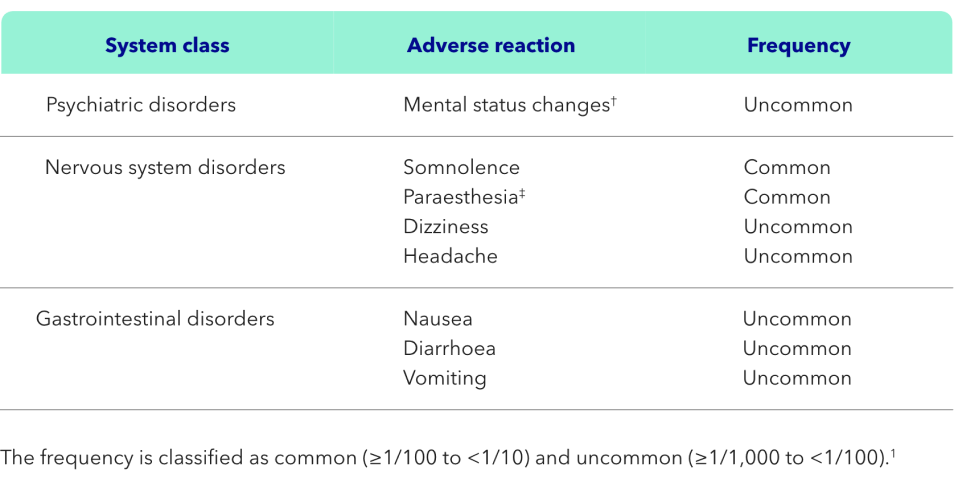
†Mental status changes included MedDRA-preferred terms of confusional state and mental status changes.1
‡Paraesthesia included MedDRA-preferred terms of paraesthesia, hypoesthesia, paraesthesia oral and hypoesthesia oral.1
Special warnings and precautions for use of KAPRUVIA®

There are no or a limited amount of data regarding the use of KAPRUVIA® in pregnant women. Animal studies do not indicate direct or indirect harmful effects with respect to reproductive toxicity. As a precautionary measure, it is preferable to avoid the use of KAPRUVIA® during pregnancy. It is unknown whether KAPRUVIA® is excreted in human breast milk. A risk to the newborns/infants cannot be excluded. A decision must be made whether to discontinue breastfeeding or to discontinue/abstain from KAPRUVIA® therapy taking into account the benefit of breastfeeding for the child and the benefit of therapy for the woman. Animal studies have shown excretion of KAPRUVIA® in breast milk. There are no data on the effect of KAPRUVIA® on fertility in humans. In rat studies with KAPRUVIA®, there was no effect on fertility. This medicinal product contains less than 1 mmol sodium per vial, that is to say it is essentially sodium-free.1

Hyperkalaemia frequently occurs in patients with chronic kidney disease on haemodialysis. In placebo-controlled clinical studies, a numerically higher rate of AEs of hyperkalaemia was reported for the KAPRUVIA®-treated patients (4.7%; 20/424 patients) compared with placebo (3.5%; 15/424 patients). No causal relationship was established. Frequent monitoring of potassium levels is recommended.1

Dizziness and somnolence have occurred in patients taking KAPRUVIA® and may subside over time with continued treatment. Concomitant use of sedating antihistamines, opioid analgesics or other CNS depressants may increase the likelihood of these adverse reactions and should be used with caution during treatment with KAPRUVIA®. Compared with placebo, the incidence of somnolence was higher in KAPRUVIA®-treated subjects 65 years of age and older (7.0%) than in KAPRUVIA®-treated subjects less than 65 years of age (2.8%).1

No evidence of physical dependence or adverse events related to withdrawal were seen in the phase III KALM‑1 study of KAPRUVIA® in HD patients with moderate‑to‑severe pruritus. This study only lasted 12 weeks, with a 2‑week discontinuation period, so the conclusions that can be drawn are limited.1,3

KAPRUVIA® is a peripherally acting kappa opioid receptor agonist with restricted access to the CNS. The blood-brain barrier (BBB) integrity is important for minimising KAPRUVIA® uptake into the CNS. Patients with clinically important disruptions to the BBB (e.g., primary brain malignancies, CNS metastases or other inflammatory conditions, active multiple sclerosis or advanced Alzheimer’s disease) may be at risk for KAPRUVIA® entry into the CNS. KAPRUVIA® should be prescribed with caution in such patients, taking into account their individual benefit-risk balance, with observation for potential CNS effects.1

KAPRUVIA® has not been studied in patients with New York Heart Association class IV heart failure. In the pivotal clinical studies a small numerical imbalance of cardiac failure and atrial fibrillation events was observed in the KAPRUVIA® treated patients compared to placebo, in particular among patients with a medical history of atrial fibrillation who discontinued or missed their atrial fibrillation treatment. No causal relationship was established.1
KAPRUVIA® has a low likelihood of drug-drug interactions1
Data from clinical and preclinical studies showed that KAPRUVIA®:1

exhibits low-to-moderate plasma protein binding (24%-32%), which limits the potential for displacement of other highly protein-bound drugs.

is not a substrate or inhibitor of major drug transporters, and has minimal to no potential to induce major cytochrome P450 enzymes, limiting the potential for drug‑drug interactions.
is predominantly excreted unchanged by the kidney in healthy subjects and eliminated predominantly unchanged via faeces in patients on HD with renal impairment.
References & footnotes
Footnotes
AE, adverse event; BBB, blood-brain barrier; CKD-aP, chronic kidney disease-associated Pruritus; CNS, central nervous system; HD, haemodialysis; IV, intravenous; SAE, serious adverse event; TEAE, treatment-emergent adverse event.
References
- KAPRUVIA® Summary of Product Characteristics. Available at: www.medicines.org.uk.
- Wiener D, et al. Kidney Med. 2022;24(10):100542.
- Fishbane S, et al. N Engl J Med 2020;382(3):222-232.
▼ This medicinal product is subject to additional monitoring. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions.
UK-DFK-2300071 (v2.0) | Date of preparation: March 2025
Safety & Efficacy
Safety and effectiveness of KAPRUVIA® (difelikefalin) in patients with moderate‑to‑severe Pruritus undergoing haemodialysis (HD): an open-label, multicentre study2
Key learning points
- The primary objective of the open-label multicentre study was the safety of KAPRUVIA® assessed through monitoring of adverse events (AEs), vital signs, 12-lead electrocordiogram (ECG) and laboratory values2
- KAPRUVIA® was generally well tolerated, with no serious treatment-related treatment-emergent adverse events (TEAEs) (primary endpoint)2
- 66% (128/194) of patients on KAPRUVIA® achieved ≥3-point improvement from baseline in sleep quality score at Week 12 (secondary endpoint)2
Safety of KAPRUVIA® was assessed through monitoring of AEs, vital signs, 12-lead ECG and laboratory values2
In this open-label, multicentre study, 222 patients between the ages of 18 and 85 with CKD-associated Pruritus (CKD-aP) on HD received 0.5 micrograms/kg of intravenous KAPRUVIA® three times weekly for 12 weeks.2
Study design and primary and secondary endpoints


KAPRUVIA® was generally well tolerated, with no serious treatment-related treatment-emergent adverse events (TEAEs) (primary endpoint)2
The safety of KAPRUVIA® was monitored through AEs, vital signs, 12-lead ECG, and clinical laboratory tests through to Week 12 (primary endpoint).2
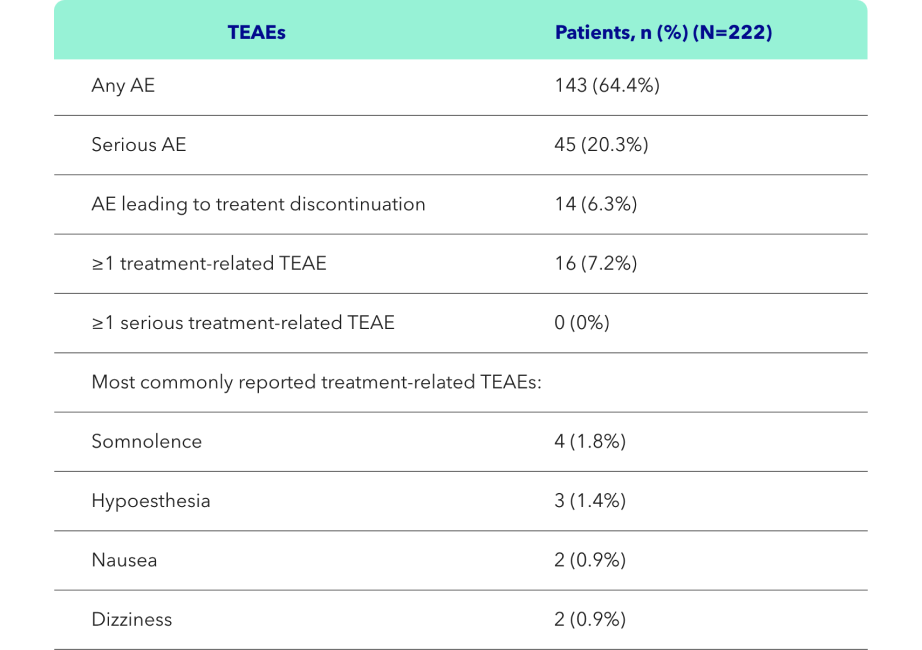

Figure adapted from Weiner D, et al. 20222
Effect of KAPRUVIA® on itch intensity and sleep quality2
Proportion of patients achieving at least a 3-point improvement from baseline in WI-NRS at 12 weeks:2*
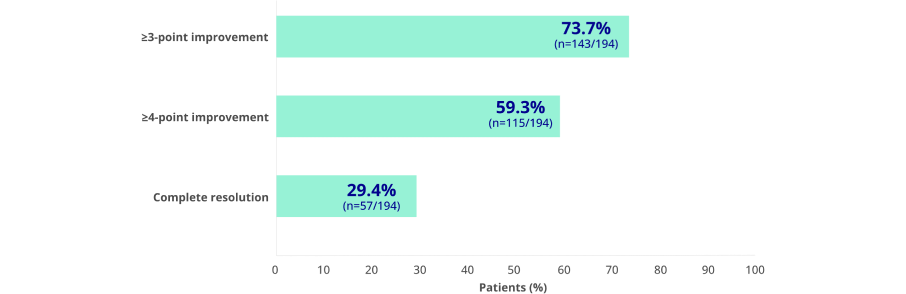
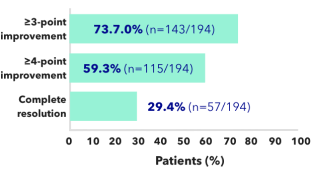
Figure adapted from Weiner D, et al. 20222
*Complete resolution defined as ≥75% of weekly mean WI-NRS scores equal to 0 or 1.
Proportion of patients achieving at least a 3-point improvement from baseline in sleep quality score at 12 weeks:1†
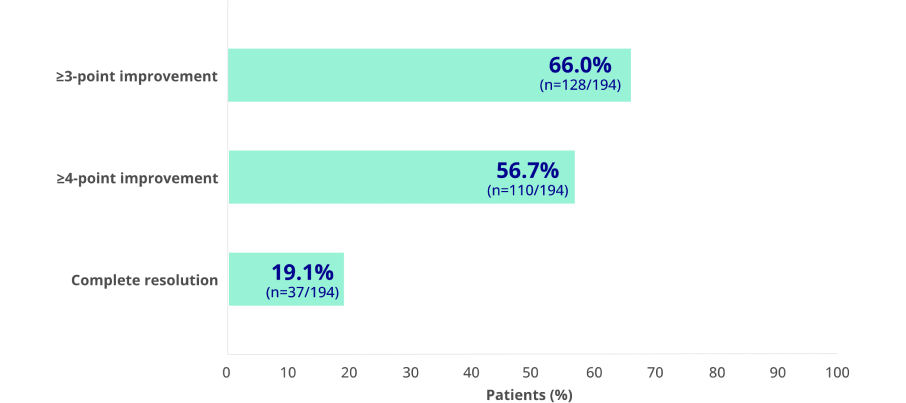
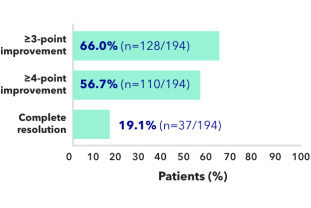
Figure adapted from Weiner D, et al. 20221
†Sleep Quality Questionnaire total score assessments (range of possible scores, 0 [did not interfere] to 10 [completely interfered]) including ≥3-point and ≥4-point improvement in weekly mean score and complete resolution (all scores equal to 0) at Week 12. During the 1-week run-in period and at baseline, 2.7% of patients had all Sleep Quality scores equal to 0.1
Study limitations: the majority of participants were enrolled in the US (91%), 49% were black or African American, and all participants were required to have adequate dialysis; the results may not be generalisable to other populations. This was a single-arm, open-label trial design with no placebo control group. A minimum clinically meaningful change for the Sleep Quality NRS has not yet been established for this patient population; however, the substantial improvements reported suggest that these changes are likely to be of clinical relevance.2
References & footnotes
Footnotes
AE, adverse event; CKD-aP, CKD-associated Pruritus; ECG, electrocardiogram; NRS, numerical rating scale; TEAE, treatment-emergent adverse event; WI-NRS, Worst-Itch Numerical Rating Scale; IV, intravenous; QoL, quality of life.
References
- KAPRUVIA® Summary of Product Characteristics. Available at: www.medicines.org.uk.
- Weiner D, et al. Kidney Med 2022;4(10):100542.
▼ This medicinal product is subject to additional monitoring. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions.
UK-DFK-2300072 (v2.0) | Date of preparation: March 2025
NICE
KAPRUVIA® (difelikefalin) is recommended by NICE2
Key learning points
- KAPRUVIA® is recommended by the National Institute for Health and Care Excellence (NICE)1
- KAPRUVIA® is directly commissioned by NHS England, meaning that funding is outside of the dialysis tariff
- KAPRUVIA® is available via the Blueteq funding process with five simple yes/no questions
How is KAPRUVIA® funded in England?
- KAPRUVIA® is directly commissioned by NHS England
- KAPRUVIA® is not in the dialysis drug tariff
- For a trust to have KAPRUVIA® funded by NHS England, a Blueteq form must be filled out first. This is a simple process that takes just a few minutes
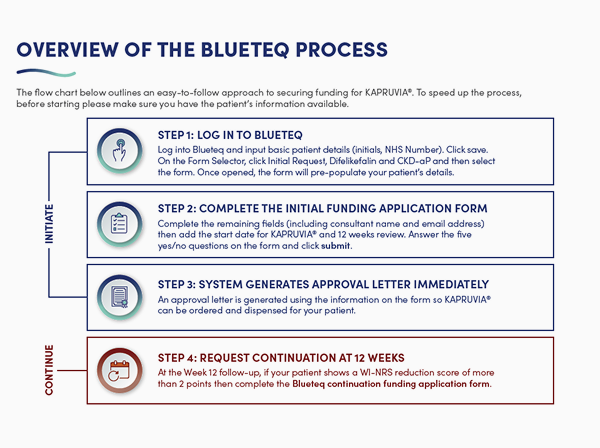
Overview of the Blueteq process
The flowchart below outlines an easy-to-follow approach to securing funding for KAPRUVIA®.
Initiate

Step 1: Log in to Blueteq
- Log into Blueteq and input basic patient details (initials, NHS number). Click save
- On the Form Selector, click Initial Request, Difelikefalin and CKD-aP, and then select the form. Once opened, the form will pre-populate your patient’s details

Step 2: Complete the initial funding application form
- Complete the remaining fields (including consultant name and email address), then add the start date for KAPRUVIA® and 12 weeks review
- Answer the five yes/no questions on the form and click submit
- I confirm that the patient is an adult with moderate-to-severe pruritus associated with kidney disease
- I confirm the patient is having in-centre haemodialysis
- I confirm the patient has pruritus despite best supportive care
- I confirm the patient will receive difelikefalin in accordance with its marketing authorisation (note the Summary of Product Characteristics states that no more than four doses per week should be administered even if the number of haemodialysis treatments in a week exceeds four)
- I confirm that the stopping criteria have been explained and agreed with the patient/carer before the treatment is started

Step 3: System generates approval letter immediately
- An approval letter is generated using the information on the form so KAPRUVIA® can be ordered and dispensed for your patient
Continue

Step 4: Request continuation at 12 Weeks
- After 12 weeks of treatment, a Blueteq continuation request is required to enable your patient to continue taking KAPRUVIA® if they have had an acceptable reduction in itch, defined as a WI-NRS score reduction of more than 2. No further forms are required
- Please follow Steps 1–3 as for the initiation form, except:
- On the Form Selector in Step 1, choose Continuation Request to bring up the correct document
- In Step 2, answer the three yes/no statements on the form and click submit
- I confirm that the patient remains eligible for difelikefalin (as described in NICE TA890)
- I confirm there is a sufficient reduction in itch during the first 12 weeks of treatment; a change in WI-NRS of more than 2 is considered a sufficient reduction
- I confirm the patient will continue to receive difelikefalin in accordance with its marketing authorisation
Watch Dr Kieran McCafferty give an overview of the Blueteq process at UK Kidney Week 2024
Time to watch: 2 mins 31 secs
Information correct at time of recording
References & footnotes
Footnotes
CKD-aP, chronic kidney disease-associated pruritus; CKD, chronic kidney disease; HD, haemodialysis; NHS, National Health Service; NICE, National Centre for Health and Care Excellence; SMC, Scottish Medicines Consortium; UK, United Kingdom; WI-NRS, Worst-Itch Numerical Rating Scale.
References
- Kapruvia SmPC.
- NICE (2023). Difelikefalin for treating pruritus in people having haemodialysis. Available at: https://www.nice.org.uk/guidance/TA890. Date accessed: June 2025.
▼ This medicinal product is subject to additional monitoring. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions.
UK-DFK-2400160 | Date of preparation: June 2025
Resources







References & footnotes
Footnotes
CKD-aP, chronic kidney disease-associated pruritus; CKD, chronic kidney disease; HD, haemodialysis; WI-NRS, Worst Itch-Numerical Rating Scale.
References
- Kapruvia SmPC.
▼ This medicinal product is subject to additional monitoring. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions.
UK-DFK-2300060 (v6.0) | Date of preparation: July 2025
NICE
KAPRUVIA® (difelikefalin) is recommended by NICE2
Key learning points
- KAPRUVIA® is recommended by the National Institute for Health and Care Excellence (NICE)1
- KAPRUVIA® is directly commissioned by NHS England, meaning that funding is outside of the dialysis tariff
- KAPRUVIA® is available via the Blueteq funding process with five simple yes/no questions
How is KAPRUVIA® funded in England?
- KAPRUVIA® is directly commissioned by NHS England
- KAPRUVIA® is not in the dialysis drug tariff
- For a trust to have KAPRUVIA® funded by NHS England, a Blueteq form must be filled out first. This is a simple process that takes just a few minutes
Overview of the Blueteq process
The flowchart below outlines an easy-to-follow approach to securing funding for KAPRUVIA®.
Initiate
Step 1: Log in to Blueteq
- Log into Blueteq and input basic patient details (initials, NHS number). Click save
- On the Form Selector, click Initial Request, Difelikefalin and CKD-aP, and then select the form. Once opened, the form will pre-populate your patient’s details
Step 2: Complete the initial funding application form
- Complete the remaining fields (including consultant name and email address), then add the start date for KAPRUVIA® and 12 weeks review
- Answer the five yes/no questions on the form and click submit
- I confirm that the patient is an adult with moderate-to-severe pruritus associated with kidney disease
- I confirm the patient is having in-centre haemodialysis
- I confirm the patient has pruritus despite best supportive care
- I confirm the patient will receive difelikefalin in accordance with its marketing authorisation (note the Summary of Product Characteristics states that no more than four doses per week should be administered even if the number of haemodialysis treatments in a week exceeds four)
- I confirm that the stopping criteria have been explained and agreed with the patient/carer before the treatment is started
Step 3: System generates approval letter immediately
- An approval letter is generated using the information on the form so KAPRUVIA® can be ordered and dispensed for your patient
Continue
Step 4: Request continuation at 12 Weeks
- After 12 weeks of treatment, a Blueteq continuation request is required to enable your patient to continue taking KAPRUVIA® if they have had an acceptable reduction in itch, defined as a WI-NRS score reduction of more than 2. No further forms are required
- Please follow Steps 1–3 as for the initiation form, except:
- On the Form Selector in Step 1, choose Continuation Request to bring up the correct document
- In Step 2, answer the three yes/no statements on the form and click submit
- I confirm that the patient remains eligible for difelikefalin (as described in NICE TA890)
- I confirm there is a sufficient reduction in itch during the first 12 weeks of treatment; a change in WI-NRS of more than 2 is considered a sufficient reduction
- I confirm the patient will continue to receive difelikefalin in accordance with its marketing authorisation
Watch Dr Kieran McCafferty give an overview of the Blueteq process at UK Kidney Week 2024
Time to watch: 2 mins 31 secs
Information correct at time of recording
References & footnotes
Footnotes
CKD-aP, chronic kidney disease-associated pruritus; CKD, chronic kidney disease; HD, haemodialysis; NHS, National Health Service; NICE, National Centre for Health and Care Excellence; SMC, Scottish Medicines Consortium; UK, United Kingdom; WI-NRS, Worst-Itch Numerical Rating Scale.
References
- Kapruvia SmPC.
- NICE (2023). Difelikefalin for treating pruritus in people having haemodialysis. Available at: https://www.nice.org.uk/guidance/TA890. Date accessed: June 2025.
▼ This medicinal product is subject to additional monitoring. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions.
UK-DFK-2400160 | Date of preparation: June 2025
SMC
KAPRUVIA® (difelikefalin) is accepted for restricted use by the SMC2
Key learning points
- KAPRUVIA® is accepted for restricted use within NHSScotland by the Scottish Medicines Consortium (SMC)2
- KAPRUVIA® has a discount Patient Access Scheme (PAS) across the UK2
SMC acceptance for KAPRUVIA®:2
KAPRUVIA® is accepted for restricted use within NHSScotland for the treatment of moderate-to-severe pruritus associated with CKD in adult patients on haemodialysis.
SMC restriction: for use in patients with an inadequate response to best supportive care for reducing itch.2
KAPRUVIA®, compared with placebo, improved itch for a greater proportion of patients with moderate-to-severe itch who were undergoing HD for end‑stage renal disease.2
This advice applies only in the context of an approved NHSScotland PAS arrangement delivering the cost‑effectiveness results upon which the decision was based, or a PAS/listed price that is equivalent or lower.2
References & footnotes
Footnotes
CKD-aP, chronic kidney disease-associated pruritus; CKD, chronic kidney disease; HD, haemodialysis; PAS, Patient Access Scheme; SMC, Scottish Medicines Consortium; UK, United Kingdom.
References
- Kapruvia SmPC.
- Scottish Medicines Consortium (SMC). SMC2623. Difelikefalin (KAPRUVIA®). Available at: www.scottishmedicines.org.uk. Date accessed: June 2025.
▼ This medicinal product is subject to additional monitoring. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions.
UK-DFK-2400161 | Date of preparation: June 2025
FAQs
Frequently Asked Questions (FAQs)
Is KAPRUVIA® recommended by NICE?
NICE has recommended KAPRUVIA®, within its marketing authorisation, for treating moderate-to-severe pruritus in adults with CKD having in‑centre haemodialysis.2
KAPRUVIA® is directly commissioned by NHS England and therefore sits outside the dialysis tariff. In England, KAPRUVIA® can be requested using the Blueteq process.
See NICE tab in menu bar
How is KAPRUVIA® administered?
KAPRUVIA® is delivered as an IV bolus at the end of in-centre HD into the venous line of the dialysis circuit during rinse-back or after rinse-back.1
- When given after rinse-back, at least 10 mL of sodium chloride 9 mg/mL (0.9%) solution injection of rinse-back volume should be administered after KAPRUVIA®.
- When given during rinse-back, no additional sodium chloride 9 mg/mL (0.9%) solution is needed.
KAPRUVIA® administration should be restricted for in-centre haemodialysis only and is intended for use by healthcare professionals experienced in the diagnosis and treatment of moderate-to-severe CKD-aP. Causes of pruritus other than CKD should be excluded before initiating treatment with KAPRUVIA®.1
How many times a week should KAPRUVIA® be given?
KAPRUVIA® is administered three times per week at the end of HD during rinse-back.1
If a fourth HD treatment is performed during the same week, a fourth dose of KAPRUVIA® should be administered at the end of HD using the recommended dose.1
No more than four doses are recommended, even if the number of dialysis treatments in a week is more than four.1
Safety and efficacy of a fourth dose has not been fully established due to insufficient data.1
KAPRUVIA® administration should be restricted for in-centre haemodialysis only and is intended for use by healthcare professionals experienced in the diagnosis and treatment of moderate-to-severe CKD-aP. Causes of pruritus other than CKD should be excluded before initiating treatment with KAPRUVIA®.1
What are the most common side effects of KAPRUVIA®?

KAPRUVIA® has been shown in clinical trials to be generally well tolerated.1
The most common (≥1/100 to <1/10) adverse events attributed to KAPRUVIA® in HD patients are somnolence and paraesthesia.1
Please consult the SmPC for a full list of adverse events.
Does a treatment algorithm exist for patients with CKD‑aP on in‑centre HD?
A publication, Agarwal R et al, 2023, proposed a treatment algorithm for screening, diagnosis, assessment and treatment of patients with CKD‑aP.3
See ALGORITHM tab in menu bar
References & footnotes
Footnotes
CKD-aP, chronic kidney disease-associated pruritus; HD, haemodialysis; IV intravenous; NHS, National Health Service; NI, Northern Ireland; NICE, National Institute for Health and Care Excellence; SMC, Scottish Medicines Consortium.
References
- Kapruvia SmPC.
- NICE (2023). Difelikefalin for treating pruritus in people having haemodialysis. Available at: https://www.nice.org.uk/guidance/TA890. Date accessed: June 2025.
- Agarwal R, et al. Clin Kidney J 2023;16:30–40
▼ This medicinal product is subject to additional monitoring. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions.
UK-DFK-2400159 | Date of preparation: June 2025
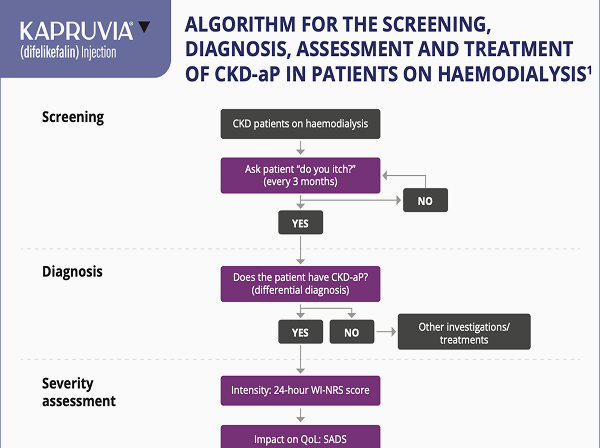
Treatment Algorithm
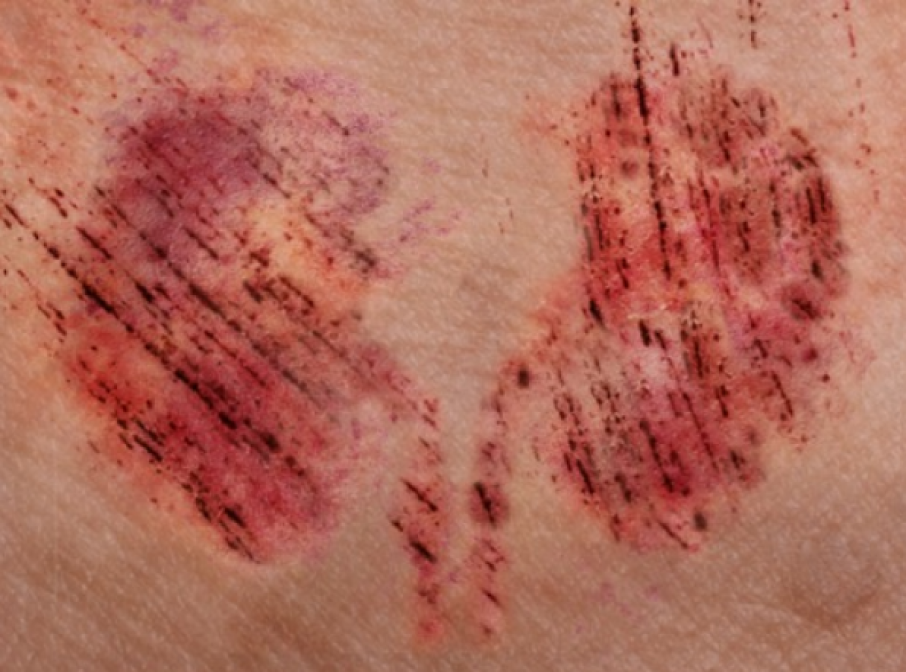
Algorithm for the screening, diagnosis, assessment and treatment of CKD-associated pruritus (CKD-aP)
Key learning points
- Ask your patients on HD: “Do you itch?”
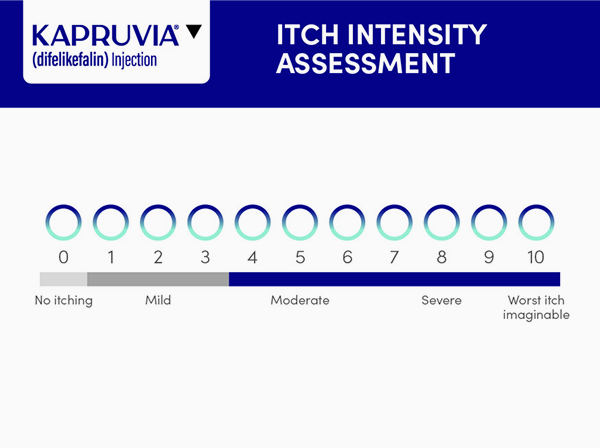
- Use the simple one-question WI-NRS scale to assess severity of itch
- A WI-NRS score of ≥4 is considered indicative of moderate-to-severe CKD-aP, where scores of 4–6 typically reflect moderate itch and scores of ≥7 denote severe itch.
- Use patient-reported measures of severity and impact of itch to direct treatment approach
A structured approach to ongoing assessment of CKD‑aP
The subjective nature of itch can often mean diagnosis and ongoing monitoring of CKD-aP is dependent on patients reporting this symptom - it is important that nephrologists and other healthcare professionals routinely and regularly ask their patients whether they are experiencing itch2
CKD-aP: screening, diagnosis, assesment and treatment
Potential treatment algorithm proposed by Agarwal R et al:3
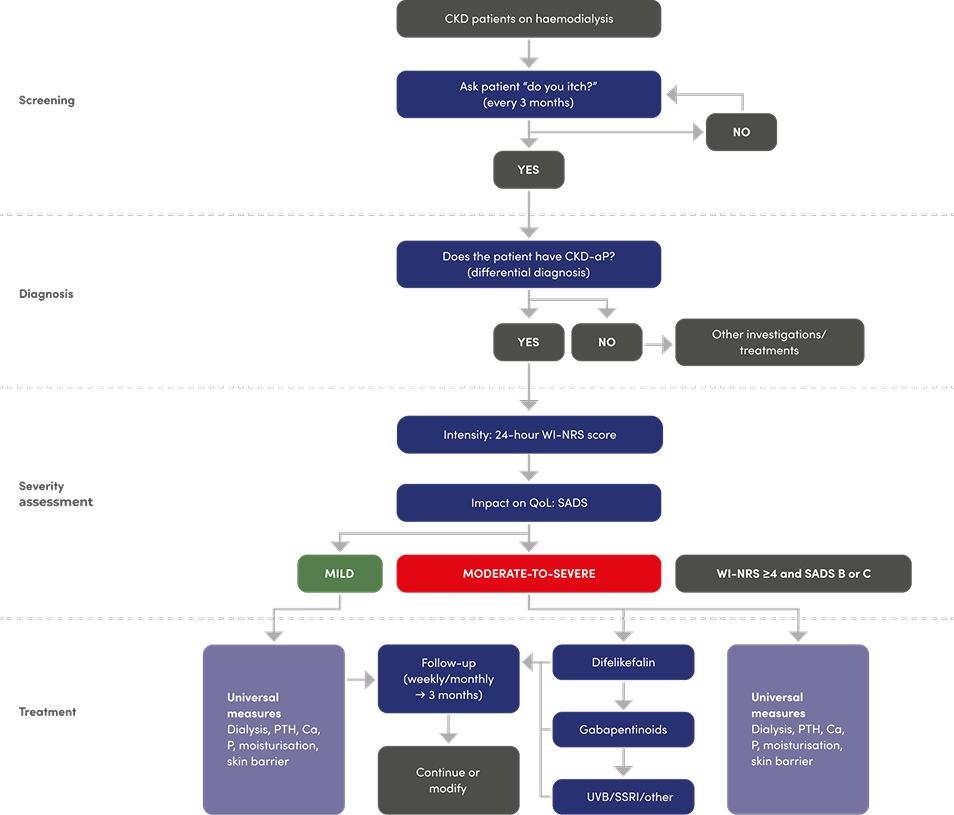
Adapted from Agarwal R et al, 20233
Please note: difelikefalin is the only licensed treatment for moderate-to-severe CKD-aP in adult patients on in-centre haemodialysis. Other pharmacological treatments in the proposed algorithm are not licensed for the treatment of CKD-aP. Please refer to the relevant Summary of Product Characteristics before making prescribing decisions.
References & footnotes
Footnotes
Ca, calcium; CKD, chronic kidney disease; CKD-aP, chronic kidney disease-associated pruritus; P, phosphorus; PTH, parathyroid hormone; QoL, quality of life; SADS, Self-Assessed Disease Severity; SSRI, selective serotonin reuptake inhibitor; UVB, ultraviolet B; WI‑NRS, Worst‑Itch Numerical Rating Scale.
References
- Kapruvia SmPC.
- Manenti L & Leuci E. Clin Kidney J. 2021;14:i8‑i15.
- Agarwal R, et al. Clin Kidney J. 2023;16:30‑40.
▼ This medicinal product is subject to additional monitoring. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions.
UK-DFK-2400163 | Date of preparation: June 2025
Adverse events should be reported. Reporting forms and information for the United Kingdom can be found at https://yellowcard.mhra.gov.uk/ or search for MHRA Yellow Card in the Google Play or Apple App Store. Adverse events should also be reported to Vifor Fresenius Medical Care Renal Pharma, care of Vifor Pharma Ltd.
Tel: +44 1276 853633. E-mail: MedicalInfo_UK@viforpharma.com.

